Unprecedented concurrent diagnosis of Angelman Syndrome and Epidermolysis Bullosa in a juvenile: Account
In a groundbreaking study published in the journal Clinical Case Reports, researchers from Barcelona have reported a rare case of a 9-month-old boy diagnosed with Angelman syndrome, epidermolysis bullosa, and autosomal recessive deafness type 57.
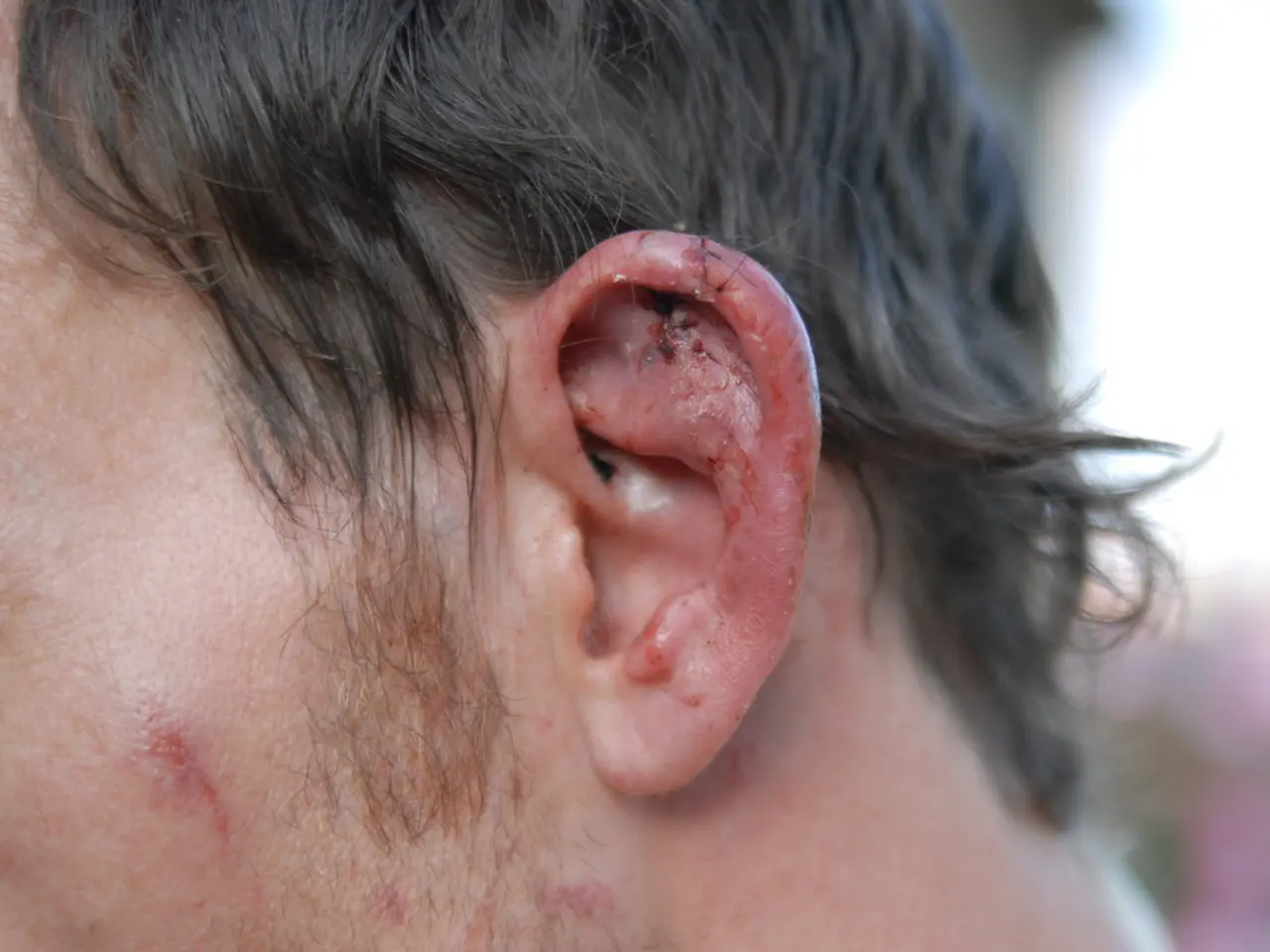
From an early age, the infant displayed global muscle weakness, struggling to sit upright by 17 months. Additionally, he exhibited facial deformities, including weak facial muscles, low-set ears, and a smaller-than-normal lower jaw. The boy also had difficulties eating and was behind other children in developing skills such as sitting up and talking, showing signs of hearing loss.
The researchers found a rare mutation in the PDZD7 gene, inherited from both the father and the mother, which may be a significant barrier to the boy's language development. This mutation, combined with a portion of chromosome 15 inherited from the mother that was found to be deleted, is the likely cause of the Angelman syndrome diagnosis. Neither parent carried this genetic defect.
The boy's case of epidermolysis bullosa is junctional EB, a moderate to severe form of the disease caused by COL17A1 gene mutations. The COL17A1 gene encodes a portion of collagen, a protein complex that gives structure and strength to connective tissues such as the skin, tendons, and ligaments. This explains the boy's fragile skin that easily blisters and tears, a characteristic of epidermolysis bullosa.
The study's findings emphasize the importance of continuously considering new signs and symptoms over time for a more accurate or even a new diagnosis. The co-existence of Angelman syndrome and epidermolysis bullosa is a rare case, estimated at one in a billion. The combined occurrence of Angelman syndrome and intermediate junctional epidermolysis bullosa would be approximately 0.97 cases per billion births.
Despite his physical challenges, the infant was very sociable and could play and interact with others. However, by 9 months, he had become uncommunicative and did not cry, babble, or make demands on his parents, which is a common symptom of Angelman syndrome.
The boy's case serves as a reminder of the importance of thorough genetic testing and the need for continued research into rare genetic disorders. The study's title is "Coexistence of junctional epidermolysis bullosa, autosomal recessive deafness type 57, and Angelman syndrome: A case report."
Read also:
- Recognition of Exceptional Patient Care: Top Staff Honored by Medical Center Board
- A continuous command instructing an entity to halts all actions, repeated numerous times.
- Oxidative Stress in Sperm Abnormalities: Impact of Reactive Oxygen Species (ROS) on Sperm Harm
- Is it possible to receive the hepatitis B vaccine more than once?







{kind=link}